What is medicinal product (drug) repurposing?
Repurposing a medicinal product (drug or biologic) involves gaining a licence for an already approved, non-patented product, in different indications from that already approved.
Why is drug repurposing important?
When a doctor prescribes a drug ‘off-label‘, it is likely that the doctor believes the drug will work in an indication for which it is not licensed. In some circumstances, the doctor could have identified a real opportunity to bring a drug to market for the benefit of those patients suffering from the indication. However, in the absence of a larger body of clinical data, the doctor would find it difficult to convince his peers of the drugs’ potential.
If the doctor were already to be sharing clinical observations with other doctors specializing in the prospective new indication, that may be a different prospect. As dialogue grows, the doctors may feel that if the drug were licensed, based on known safety and efficacy data, then every prescribing doctor could benefit from the product’s potential.
This could be a significant breakthrough in the world of medical intervention.
What would it take for doctors to repurpose drugs?
For decades, it has been assumed only pharmaceutical companies have the wherewithal to repurpose medicines. This article challenges that assumption, based on changing business models in the industry. These changes have created a skills-base of qualified contractors and contract organisations able to offer whatever is required to bring a medicinal product to market.
The message to doctors and other health professionals is that there is just one gap in their armoury—knowledge of the supply-chain that delivers a medicinal product to patients.
Surely, this can’t be true?
If you think I am stretching the truth here, please listen to this.
As the large pharmaceutical companies have grown during the blockbuster era, they have divested the facilities, skills and logistics processes required to develop a medicinal product. Today, there is a vast array of contractor companies, working to a fee-for-service business model, ready and waiting to help people develop and repurpose medicines. All anyone who wants to develop a medicine needs to know is where to go to get the help they need.
True, it will require raising and spending some funds, but for a repurposed drug, it could be a fraction of the cost to develop a new medicine from scratch.
I’m a doctor wishing to repurpose a medicinal product – what do I need to know?
Let’s assume you would like to repurpose an existing licensed medicinal product. There are broadly two classes of medicinal product involved:
* Small-molecule products – that means they are made using industrial chemistry.
* Biologic (large-molecule) products – that means they are made from living things, such as animal and human cells.
It is important to know which class of medicine you wish to repurpose, as the requirements and regulations are very different – biologic products are far more challenging to develop, manufacture and distribute than small-molecule medicines due to the nature of living things.
Historically, the large pharmaceutical companies have carried out the research and development activities (R&D) to bring a novel medicine to market, for both small-molecule and biologic products.
The products these companies develop, and market, are known as originator (or innovator or reference) products.
Copies of small-molecule originator products are known as generics.
Copies of originator biologic products are known as biosimilars.
Assuming the most straight-forward case of repurposing is a generic small-molecule medicine, you will need to know about the Regulatory Authorities involved.
Regulatory Authorities
Regulatory Authorities principally approve drugs for clinical trials and sale, inspect organisations and facilities for suitability to operate, and monitor the safety of medicines. Regulations are laws so are not to be taken lightly.
Fortunately, most Regulatory Authorities have comprehensive websites and should be your first source of reference for all regulatory matters. The FDA website is an example.
Additionally, the global organisation The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) has worked with the three major Regulatory Authorities – Food & Drug Administration (US FDA), European Medicines Agency (EU EMA) and the Pharmaceuticals and Medical Device Agency (Japan PMDA) – to harmonise certain key aspects of regulation globally.
The good news for those repurposing medicines is that the rules have been clearly spelt out.
The less good news is that you must know where to look and how to know what applies to your particular circumstances…but more good news — there are skilled and experienced regulatory affairs, and other experts, out there waiting to help.
Licencing a medicine for market
Knowledge of the major Regulatory Authority fundamentals will allow you to understand the support you will need to submit a license application. Submission of an (electronic) Common Technical Document is mandatory for applications to market a prescription-only product.
There are three modules to be submitted:
* Module 3: Chemistry (Chemistry, Manufacturing and Controls – CMC)
* Module 4: Nonclinical Study reports
* Module 5: Clinical Study Reports.
The decision to approve, or not, is based on the contents of the eCTD, together with answers to Regulatory Authority questions arising from the review.
Doctors have a head start – but there’s a handicap to overcome
This is where the news becomes really encouraging for doctors. Modules 4 & 5 will have a substantial amount of regulatory and real-world data (RWD) and real-world evidence (RWE) behind them — and RWD/RWE are increasingly being pursued by Regulatory Authorities as a source of information to support decision-making.
Doctors are constantly developing RWD/RWE in their day-to-day work treating patients and that has brought this to the fore in more recent years. It would be a relatively short step to start collecting and formatting the data for Regulatory Authorities to review, especially if it were for repurposing a long-standing generic medicine.
That’s the head-start, what is the handicap?
Module 3 (see bottom of Figure 1 below) is unchartered territory for doctors. It relates to the chemistry, manufacturing and controls (CMC) aspect of developing a drug (sometimes referred to as just the Chemistry, or Quality Module).
This section of the eCTD filing is where all the details of suppliers, manufacturers, material and product specifications, test procedures, development protocols, and other data specified by the Regulatory Authority must be detailed. It is, in effect, the supply-chain, from beginning to end, soup to nuts, or source to destination.
The handicap is that none of these aspects are covered in schools of learning for those with ambitions of forging a career in medicine or the medicines industry. In fact, CMC aspects don’t find their way into very many, if any, schools of medical learning.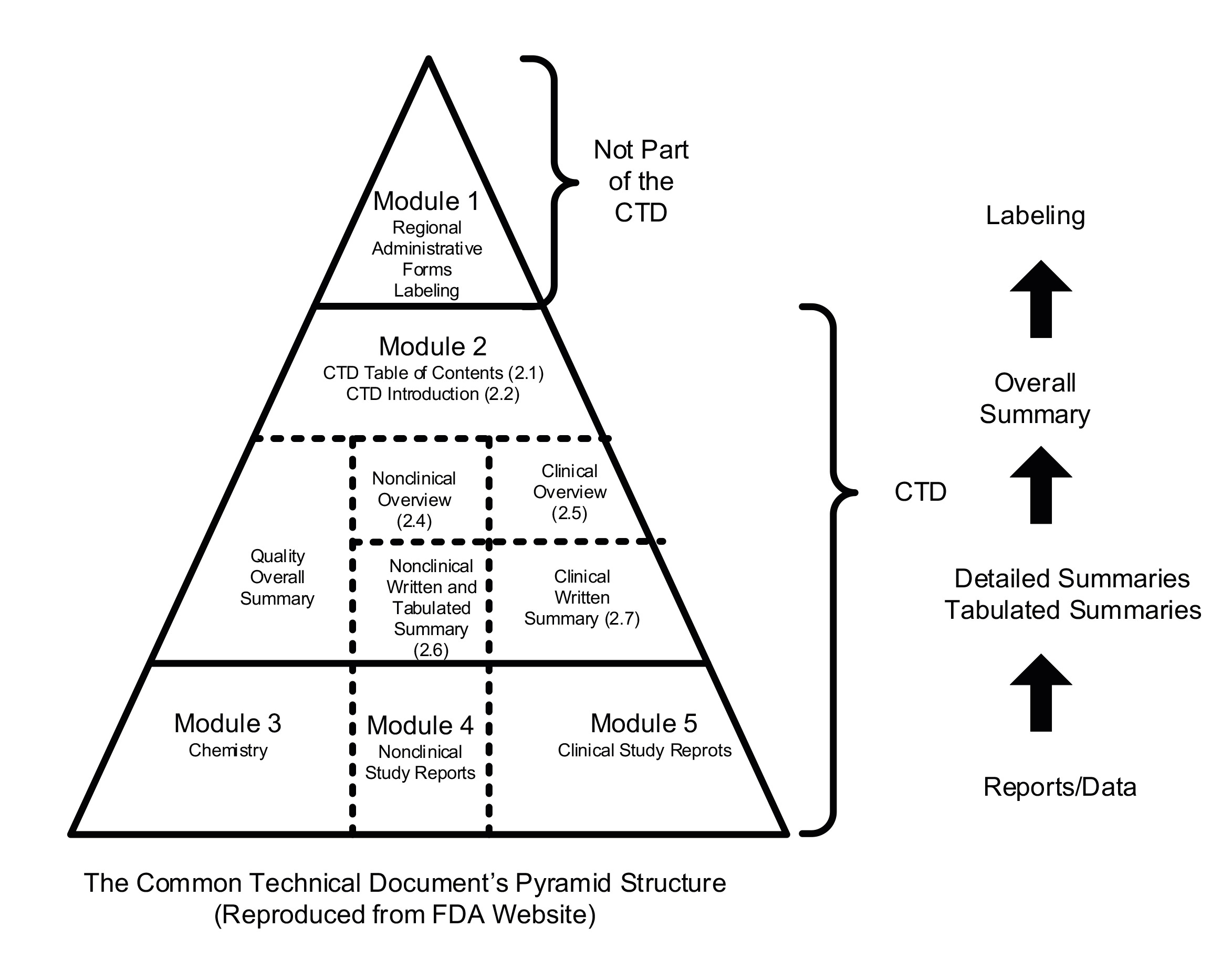
Figure 1: Common Technical Document (eCTD)
How to overcome the handicap
Lao Tzu is often quoted thus “every journey begins with a single step.”
You have started your journey here by reading this article. You may decide that is far enough for you, and who would blame you, it could be tough going. Hopefully though, you might consider taking a second step, by looking up some of the topics and references covered above.
For more on this topic and a much broader perspective, Transforming the Pharmaceutical Supply Chain will be published by Wiley on September 17, 2025, providing in-depth knowledge for doctors and other professionals involved with health, medicinal products, and patient wellbeing.
This is the product description:
“Effective and insightful solutions to the most pressing supply chain challenges facing pharmaceutical companies today.”